|
������������RDS��Rh������Ϊ����RP������صĻ���RP��Rh����ͻ���ڹ������н϶��о���������RP��RDS����ͻ���ڹ�����δ�����档���Ƕ�ͬһ��ϵ��2��RP���߽��в����������ʾRDS����216�����2�������������C��Tͻ�䣨Pro216 Leu��������������ͻ�����Ӧ���۲����ͣ�̽��RDS����Pro216 Leu ͻ����RP�۲����͵Ĺ�����RDS����ͻ�䵼��RP�ķ������ơ�
����1.RDS����ͻ�����۲����͵Ĺ�����RP��һ���Ŵ������Ժ��ٴ������Խ�ǿ�ļ������Ŵ������Գ�����Ϊ�Ŵ���ʽ�Ķ������⣬������Ϊ�ж��ֻ���ͻ����ڣ��ٴ����������Ϊ����������ͬ��ϵ��RP�ٴ����ָ�����ͬ��
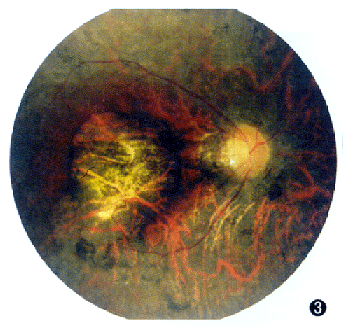
����Rishman����5������2����RDS����Pro216 Serͻ�������RP��ϵ���۲����ͣ�����һ����ϵ��4��RP���߾���RDS����Pro216 Serͻ�䣨216�����1λ������C��T������20��30��ʱ����ҹä֢״��30������������ʼ�½�������ܱ���Ұ��խ���۵ı�Ϊ��ɢ��RP������������Ĥή��������2���лư�������ή������(atrophic-appearing foveal lesion)��1���ɼ��ư߲�����ĤǰĤ��1���лư߲�ɫ�س��š���һ��ϵ����֤��Ҳ��RDS����Pro216 Serͻ�䣬39��ʱ����ҹä���۵��ɼ����͵�����Ĥή����ư�������ή����������Ĥ��������ɢ�ڵĹ�ϸ����ɫ�س��š����Ƿ����й���RP������RDS����Pro216 Leuͻ�䣨����֤ʵ216�����2λ������C��T�����۲�������20��ʱ����ҹä��������ʼ�½�����״����в�ͬ�̶ȵĻ��ǣ�����ɫ��������ĤѪ����խ�������������������ϡ���ϸ����ɫ�س��ź�����Ĥή����1���ư߲�������������״����Ĥ����Ĥή����1����״����������ư߲���F-ERG�����ʾ����Ĥ���塢����ʷ�ӦϨ���ͣ���ҰΪ�������ӵ��������������������ɴ˿ɼ�RDS����216������ͻ����۲����ʹ���Ϊ��ɢ��RP��ư߲����䡣
��������������״����Ĥή���ĵ����۵ı�Ϊ�ư����߽����������״����Ĥ��ɫ����Ƥ������ĤëϸѪ��ή������FFAʾ�ư�������ĤëϸѪ����ע����ӫ���Ե����ǿӫ����������ڻư����ʰߵ�״��ӫ�⡣�����е���֤�߳���˫�۶ԳƵ�����������״����Ĥ����Ĥή���⣬����������ɫ�䵭������ĤѪ�ܱ�ϸ���������������Ĥ�ɼ����͵Ĺ�ϸ����ɫ�س��ţ�F-ERGҲ��ʾ���弰�巴Ӧ��ʧ��˵����RDS����Pro216 Leuͻ����۲�����Ϊ��ɢ��RP������������״����Ĥ����Ĥή����
����2.RDS����ͻ����RP�ķ��ӷ������ƣ���RDS������2���ں��Ӻ�3�������ӣ������ڹ�������е��������T��Ĥ��Ե���ף�������ϵ������������������Ĥ��Ե����Ľṹ�ǵ��ף���С��RDS���ײ����ͬԴ���Ĥ��Ե����ά�ָ��������Ĥ�ṹ���ȶ���������Ҫ������6,7����
����Travis ����7����RDS��cDNA����RFLP����������һЩRP��ϵ��RDS����������8����RDS����ͻ������С������Ĥ���ԵIJ����ı�Ϊ���ڸ�����������������쳣��ǿ����֮��ʼ�����ر��ԣ�������������RPʮ�����ơ�С��������Ŵ�������Ĥ������RDS����ͻ�����ϵʹRDS�����ΪRP����Ҫ��ػ�����RDS������������صĵ����λ���λ����Ĥ��Ե��������Ĥ���϶��2��ת������216�������İ����ἴλ�ڴ˴����������ڻ��������ϸ߶ȱ��أ�Pro216 Leuͻ����ܽ�Ӱ����Ĥ��Ե���Ķ����ṹ������ص��Ľ�ϣ��Ӷ�����RP�ķ���������Ŀǰ�Ѳ��RDS����ͻ����RP�ķ��ӷ����й���4������RDS������RP�ķ��ӷ��������в�ʮ���������δ��ͻ��RDS�����ת����ϸ����ת��������о������������о�����Ǵ�RDSС��ĸ��ַ����л�ã��й�ȷ�з���������������̽�֡�

ͼ1������RDS����������2��RGR�����Ӿͼ
ͼ2��RDS�����Pro216Leuͻ��ø������Ӿͼ
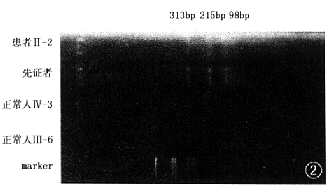
ͼ3����֤�ߵIJ�ɫ�۵�ͼ��
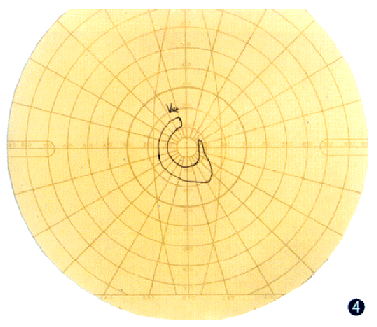
ͼ4����֤�ߵ�Coldmann��Ұͼ
��������飺���룬��ʿ��
���������
����1��Gilbert C, Rahi J, Eckstein M, et al. Hereditary disease as a cause of childhood blindness: regional variation. Ophthalmic Genetics, 1995, 16:1-10.
����2��Sambrook J, Frish EF, Manistis T. Molecular cloning: a laboratory manual. 2nd ed. New York: Cold Spring Harbor Laboratory, 1989. 463-690.
����3��Travis GH, Christerson L, Danielson PE, et al. The human retinal degeneration slow (RDS) gene: chromosome assignment and structure of mRNA. Genomics, 1991, 10:733-739.
����4��Wells J, Wroblewski J, Keen J, et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nature Genet, 1993,3:213-218.
����5��Fishman G, Stone E, Gilbert LD, et al. Clinical features of a previously undescribed codon 216 (proline to serine) mutation in the peripherin /retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Ophthalmology, 1994, 101:1409-1421.
����6��Connell G, Basscom R, Molday L, et al. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc Natl Acad Sci U S A, 1991, 88:723-726.
����7��Travis GH, Brennan MB, Danielson PE, et al. Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (RDS). Nature, 1989, 338:70-73.
����8��Farrar GJ, Jordan SA, Kenna P, et al. Autosomal dominant retinitis pigmentosa: localization of a disease gene (RP6) to the short arm of chromosome 6. Genomics, 1991, 11:870-874.
���ո����ڣ�1999-05-21�� ��һҳ [1] [2] [3] |



