����1���������ԣ���������쳣2������Ժ�����ߵ��ٴ����ֽ�ϲ���֧��IgA����(IgAN)�������Ե���С�ܼ���������ԭ����IgAN���͡���һ����Ѱ���¼̷���IgAN�Ļ����������ۿƼ����ʾ��ĤK-F������ϻ��ߵ��ۡ��Ρ��Բ����䣬���ΪWilson����
��������ժҪ
��������,���ԣ�19�꣬��������쳣2���࣬��2009��2��2����Ժ��
�����ֲ�ʷ 2006��6��15�ջ����ڽ�ʳ����ʳ�����ֶ��ġ�Ż�£�Ż����Ϊθ������鷢��(����38.5�棬)����ʹ���������»ָ�����������Ż�º�ת���ڵ���ҽԺ���棺����2+����Ѫ3+��Ѫѹ��Ѫ�弡��(Scr)������6��19�ջ�������Ժ����������ʾ��0.71 g/24h�����ϸ��60��/ml����һ�ͣ�Scr 1.03 mg/dl�� B��˫����С����̬�������Ժ��ײ�����0.43��0.85 g/24h�����ϸ��1��50��/ml�������ͣ�Scr������
����2009��1��21�ջ����ٴ���Ժ���������ʾ����������(0.79 g/24h)�� Scr ������������Ѫ֢(97 ��mol/L)��Ϊ��һ��������Ժ�����߲��������ס�����Ѫ��ҹ�����ࡢ��Ѫѹ�����������������ȡ�Ƥ��ѷ�����ǻ���ڸɡ��۸ɡ���ʹ���ڱ㼰�����½���
��������ʷ �����⡣ ����ʷ ��ĸ�彡���ǽ���飬����Ϊ���ӣ����ϼ����������Ƽ������ߣ����ϼ����Ŵ���ʷ��
������Ժ���� ����36.5�棬����76��/�֣�Ѫѹ 120/80 mmHg������18��/�֣�����ָ��(BMI) 18.8 kg/m2��������������־������ġ��Ρ������δ�����쳣��
������Ͼ���
������С�ܹ�����������
������Ժ����Һ���ʾ����������(0.67 g/24h)����������Ѫ��(���ϸ��7��13��/ml��������)��ͬʱ���ֽ�����С�ܹ�������������Ϊ���ӻƴ���ϵ���(RBP) ��������(42.88 mg/L������ֵ<0.5 mg/L)����������(508 mg/24h)��С����(������<6�������)��ռ36.0%��ѪҺ���Ҳ��ʾ��С�ܹ������ˣ�����Ϊ������Ѫ֢(99 ��mol/L)���ͼ�Ѫ֢(3.37 mmol/L)��
����
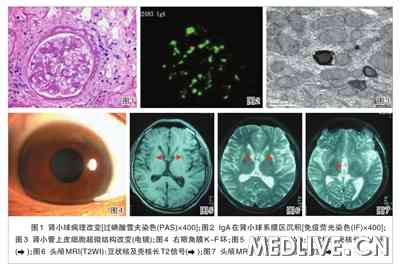
����������С�ܼ��ʲ������Ե�����ԭ������С�������ͣ���һ�������������ȷ��ϡ��⾵��21����С��1�����Է�������С��ڶ�ϵĤ����������������ұڽڶ����ֲ㣬������ά��;С������С��ή�����϶��ή��С�ܻ�Ĥ����������ɢ�ڵ����˼�����ϸ������(ͼ1)������ӫ�⣺IgG++��IgA++��C3++�������ֲ����ʿ���״������ϵĤ��(ͼ2)��IgM��C4��C1q���ԡ��羵��δ����С����С�ܻ�Ĥ��������С����Ƥϸ����������������ԣ�����Ϊ����Ĥ���룬�ձ�̡���ʧ����ø�����࣬����������С��(ͼ3)��
�������ֽ�ĤK-F��
�������ߵ��ٴ����ֽ�ϲ���֧��IgA����(IgAN)����ԭ����IgAN��С�ܼ��ʲ��������ڼ������ڣ����ٴ������벡���ı������С�����Լ��ᣬ�����������Ե���С�ܼ���������ԭ����IgAN���͡���һ��������Ѱ���¼̷���IgAN�Ļ���������������鷢�ֶ��������������ԣ�Ѫ�岹��������Ѫ�帱���δ���쳣��˫����ֽ��������Ĥ����ʱ������������֯���ͽ�ԭ��3����5��Ⱦɫ��������ʪ�Լ������µļ̷���IgAN��������IgAN�鸱��Ѫ֢�����Ŵ�������Ҳ�������� �ۿƼ����ʾ��ĤK-F��(ͼ4)����ʾ���ų��ζ�״�˱���(Wilson��)��Wilson������ͨ��IgA������֯������ͭ��л�쳣����IgAN�ͼ��������ס�
����ͷ�Ź���(MRI)����˫�ඹ״�˼��Ǻ˿ɼ���T1��T2�źţ�������������(Լ13 mm)(ͼ5-7)��B�����ʾ���������ǿ�������л��߸ι���������Ѫ��ͭ������267.1 mg/L(����ֵ110��500 mg/L)�� ��ϻ��ߵ��ۡ��Ρ��Բ����䣬���ܻ��߸ι�����Ѫ��ͭ��������������Ҳ��ȷ���Ϊ��Wilson����Wilson��������������Լ���������(��л�����), IgAN(�̷���)���������ȷ������п�Ƽ���ͭ���ƣ�ͬʱ���Զ����IJ��Ƽ����ơ� Wilson���ۼ������ټ���������쳣Ϊ��֢״����Ϊ�ټ�������������©�������������ۡ��Բ��䣬��ȱ�����͵ĸ��ಡ�䣬Ѫ��ͭ�������������ٴ���Ҳ����������©�������Ժ���鷢�����Ե���С�ܼ������ˣ���һ�����ۿƼ�飬�Ӷ���ȷ������Wilson������ϣ�Ϊ�������ƣ�Ԥ��������һ���Ρ��ԡ�����Ӯ���˱����ʱ�䡣
������������
����������Wilson�������
����Wilson�����ټ����Ŵ��Լ�����������Ϊ1/30���ڲ�ͬ���塢���弰����䣬�ٴ����ֲ���ܴ�,����ʽ�ɼ��ɻ�,�ٴ����ָ��Ӷ��,����������ⱨ�������ʴ�28%���������ױ��������ʸߴ�45%��50%�����ں���Դ�Ƚ���Wilson����֢״���ֺ�2������ҽԺ����,ʱ�䳬��3�����Դ�����ϻ��в�����ȷ�����,����Ϊ�������ϲ�������1011�������о�����������51.04%��������,19.09%������ϲ�������29.87%������֢״����3�����ڵõ���ȷ��ϡ�
����������Wilson�����ٴ���ʱ��ȷ��ϴ��������ս��Ϊ�����һ�ٴ����⣬2001���ڵ¹�������(Lpeizig)�ڰ˽����Wilson�����ſ�˹�� (Menkes��)�����ϣ�ר�ҶԲ�����Wilson����������Lpeizig����ϵͳ������4�֣���������ȷ���(��A13���1)��2007��Ala�������Wilson�����˼·��2008�������β�ѧ����ٴ��Ը��ಡ���(��)�����쳣���ֵ�Wilson����Ͻ����˹淶���Բ�����Wilson����ϲ���δ���ġ�
��������������ʾ��Wilson�����������ʼ����������ಡ��Ϊ��֢״���ټ������ͺ��ȱ����80��Wilson��������,����������ռ40% ,������ı�Ϊ�������߽�ռ3.99%������ȱ��棬24��Wilson�������а�������8�������н�1�������ಡ��Ϊ��֢״�������ಡ��Ϊ��֢״��Wilson������©�
����Wilson�������ٴ�����
����Wilson��Դ��WD ����ͻ�䲻����Ч�������������ͭ��л�ϰ���������ͭ�����ڸΡ��Ե���֯,������Ӧ���䡣����ͭҲ���Գ��������࣬���������ʼ����س̶ȽϸΡ������Լ��ᡣ
����Ϊ��Wilson�������ķ���Ƶ�ʼ����س̶�Զ����������?���벻ͬ��֯ͭת��ATPø(ATPases)�ֲ���ͬ�йء�ATP7A����С�ܻ���Ĥͭת��������Ҫ���ã����ܱ�����������ͭ���ɹ��ȵ�Ӱ�졣
�����ҹ�ѧ�ߵĹ���������Wilson���������ټ�����Ҫ������С�ܹ����������ٴ�����Ϊ��С�����ж��������������������;Ҳ���Գ�����С������Ҫ�е������Ѫ����Ѫ����������������ʽ;�������ױ�����Գ�������ʯ��
����Wilson�����IJ����ı�
����Ŀǰ��Ϊ���鵼Wilson�������ܻ��������¼��㡣��ͭ�������������Ҫ�����ڽ�����С�ܼ�Զ����С����Ƥϸ��,��С����Ƥϸ����ƽ��������Ĥ����,��С�������չ����½�����IgA������֯�������鵼�̷���IgAN����Ҫ�̷���Wilson�������������Ĥ�������Ϲ�������;ͭҲ���Խ鵼��С����,����Ѫ�۹������ɣ����״�л���ң���������ϵ��ʯ��
��������
����Wilson�����Ը������ϵͳ��Ϊ�ٴ������ij�Ⱦɫ�������Ŵ��Լ��������ҹ���Wilson����������������ټ����������ಡ��Ϊ��֢״��ʮ���ټ�����©����ٴ�����������ԭ�����С�ܼ�����ʱ��Ӧ����Wilson�����������༲�����Գ����۲����䣬�۲��������ȷ�������ϵ���Ҫ������ |



